Phylogenien aus Genomsequenzen berechnen
Forschungsbericht (importiert) 2014 - Max Planck Institut für Evolutionsbiologie
Die Berechnung von Stammbäumen (Phylogenien) ist eine der beliebtesten Anwendungen der Bioinformatik. Dazu werden evolutionäre Abstände zwischen Nukleotid- oder Aminosäuresequenzen geschätzt und daraus die Phylogenie rekonstruiert. Allerdings ist die Abstandsschätzung zwischen langen Sequenzen aufwändig. Daher wurde eine neue Methode zur ultra-schnellen Berechnung von Abständen zwischen Genomen entwickelt.
Über den Ursprung von Phylogenien
Darwins Buch Über den Ursprung der Arten enthielt eine einzige Abbildung: einen imaginären Stammbaum, auch Phylogenie genannt, mit dessen Hilfe der Gentleman aus Down seine Theorie der Evolution illustrierte. Anstelle hypothetischer Stammbäume gibt es heute eine Fülle konkreter Phylogenien, die Zeiträume von wenigen Jahrzehnten bis hin zum Alter des Lebens selbst, also rund 4,5 Milliarden Jahre, umfassen. Phylogenien werden meistens auf der Grundlage von DNA-Sequenzen erstellt, da diese sich über große Zeiträume mit ungefähr gleichmäßiger Geschwindigkeit verändern.
Der Preis einer DNA-Sequenzierung ist zwischen 2007 und 2012 um das rund 6000-fache gefallen [1]. Das eröffnet völlig neue Anwendungen für umfassende Sequenzierungen und deren Auswertung. Zum Beispiel kann heute der Verlauf bakterieller oder viraler Epidemien genau verfolgt werden, indem das vollständige Genom jedes neu isolierten Erregerstamms sequenziert wird. Daraus wachsen Sammlungen hunderter bis tausender ähnlicher Genome, deren Beziehungen untereinander als Phylogenien zusammengefasst werden können.
, daraus berechnete Distanzmatrix (B) und aus den Distanzen berechnete Phylogenie (C).")
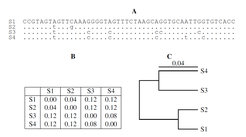
Zur Berechnung einer Phylogenie werden die Genome zunächst so untereinander geschrieben, dass sie sich möglichst wenig unterscheiden. Abbildung 1A zeigt ein solches Alignment von vier Sequenzen mit jeweils fünfzig Basen. Um die Abstände zwischen ihnen zu messen, zählt man für jedes der Paare die Unterschiede, teilt sie durch die Sequenzlänge und schreibt das Ergebnis in eine Tabelle, die sogenannte Distanzmatrix (Abb. 1B). In Abbildung 1C schließlich sind die Abstände zu einer Phylogenie zusammengefasst [2]. Dort ist zum Beispiel der Abstand zwischen S4 und S3 2 × 0,04 = 0,08, wie in Abbildung 1B notiert.
Phylogenien berechnen
Die Berechnung eines Alignments kann zeitaufwändig sein. Zum Beispiel dauert das Alignment eines Test-Datensatzes von 29 Genomen des Darmbakteriums Escherichia coli mit dem schnellen Programm mugsy [3] etwa fünfeinhalb Stunden. E. coli Genome sind durchschnittlich knapp fünf Millionen Basen lang, also fünfhundertmal länger als dieser Artikel. Die aus den alignierten Genomen berechneten Abstände ergeben die Phylogenie in Abbildung 2A. Zur Orientierung sind darin die Vertreter des bei molekularbiologischen Arbeiten häufig eingesetzten Stamms K-12 blau gekennzeichnet.
 basierend auf einem Alignment, das mit dem Programm mugsy [3] berechnet wurde, und (B) basierend auf Distanzen, die mit dem neuen Programm andi berechnet wurden. Blau dargestellt sind vier Varianten des in der Molekularbiologie sehr häufig verwendeten K-12 Stammes; rot sind die beiden Stämme wiedergegeben, deren Position zwischen den beiden Phylogenien variieren.")
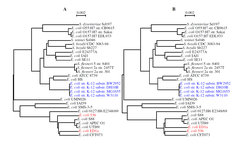
Ob fünfeinhalb Stunden für ein Alignment viel oder wenig sind, hängt von der Perspektive ab: Gemessen an dem Aufwand, die Sequenzen zu erheben, ist es kurz; gemessen an der für interaktives Arbeiten notwendigen Reaktionszeit, ist es lang. Um Daten zu analysieren, muss man verschiedene Herangehensweisen ausprobieren. Das geht aber nur, wenn jeder einzelne Rechenschritt rasch, das heißt nach Möglichkeit interaktiv, Resultate liefert. Entsprechend wünscht man sich für die Analyse von Sequenzdaten interaktive Werkzeuge. Daher entwickelte der Autor in Zusammenarbeit mit Fabian Klötzl und Peter Pfaffelhuber eine Methode, mit der die Distanzen zwischen den E. coli Sequenzen innerhalb von 19,8 Sekunden berechnet werden können [4]. Trotz dieser 1000-fachen Beschleunigung ist die daraus rekonstruierte Phylogenie in Abbildung 2B kaum zu unterscheiden von dem Alignment-Baum in Abbildung 2A.
Phylogenien aus Suffixbäumen
Eine derartige Beschleunigung bei gleichzeitiger Beibehaltung der Genauigkeit ist möglich durch die Verwendung eines neu entwickelten Distanzmaßes: Zwischen zwei Sequenzen werden möglichst lange, exakte Übereinstimmungen gesucht, die Anker heißen, da sie ähnliche Bereiche zwischen den Sequenzen miteinander verankern. Wenn ein Paar von Ankern in beiden Sequenzen den gleichen Abstand hat, werden die Unterschiede im dazwischenliegenden Bereich gezählt. Die Gesamtzahl der Unterschiede, geteilt durch die Zahl der untersuchten Basen, ergibt dann die Ankerdistanz [4].
Um solche Anker schnell zu finden, baut man für jede Genomsequenz einen Index aller darin vorkommenden Suffixe. Ein Suffix bezeichnet das Ende eines Worts, zum Beispiel ist “fix” ein Suffix von “Suffix”. Ein solcher Index heißt Suffixbaum [5] und Abbildung 3 zeigt ein Beispiel für die Sequenz CAAT. Der Baum trägt seine Wurzel oben, seine Äste sind mit Buchstaben und seine Blätter mit Zahlen beschriftet. Wenn wir die Buchstaben auf dem Weg von der Wurzel bis zu einem Blatt aneinander hängen, erhalten wir den Suffix, der an der Blattbeschriftung anfängt. Zum Beispiel ist der Pfad von der Wurzel bis zum Blatt 2 mit AAT beschriftet, dem Suffix an der zweiten Position. Um in diesem Baum etwa nach A zu suchen, fängt man bei der Wurzel an, findet in einem einzigen Suchschritt den mit A beschrifteten Ast und schaut die Anfangspositionen in den darunterliegenden Blättern nach, in unserem Fall 2 und 3. Auch im Suffixbaum eines Genoms würde diese Suche nur einen Schritt erfordern. Dank eines Suffixbaums hängt die Suche nach einem Sequenzstück also nur von dessen Länge ab, unabhängig von der Größe des Genoms.

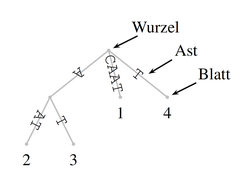
Abb. 3: Suffixbaum der Sequenz CAAT. Nähere Erläuterungen im Text.
Nun klingt eine Textsuche, deren Aufwand unabhängig von der Länge des Textes ist, kaum glaublich, stellt aber in der Tat das Funktionsprinzip jedes normalen Buchindexes dar. In der Bioinformatik sind die entsprechenden Suffixbäume dann nützlich, wenn man sie schnell konstruieren kann. Erstaunlicherweise ist die Berechnung eines Suffixbaums so einfach wie das alphabetische Sortieren aller Suffixe einer Sequenz [6, 7]. Für CAAT etwa ist das:
| Suffix | Startposition |
| AAT | 2 |
| AT | 3 |
| CAAT | 1 |
| T | 4 |
In den letzten Jahren sind zunehmend effiziente Sortierungsmethoden für Suffixe entwickelt worden [8]. Eine solche wird in andi, dem Programm zur Berechnung von Ankerdistanzen, verwendet. Wer auf einem Computer mit UNIX Betriebssystem arbeitet, zum Beispiel Mac-OSX oder Linux, kann die neuste Version des Programms hier erhalten:
https://www.github.com/evolbioinf/
und mit den dort ebenfalls hinterlegten Beispiel-Datensätzen gleich ausprobieren.
Genauigkeit und Geschwindigkeit
Ein neues Distanz-Programm muss zuallererst auf die Genauigkeit seiner Ergebnisse hin untersucht werden. Abbildung 2 zeigte bereits durch den Vergleich mit herkömmlichem Alignment, dass andi recht genau zu sein scheint. Mithilfe simulierter Daten kann man diesen Eindruck systematisieren und stellt fest, dass die Übereinstimmung zwischen Schätzung und Simulation solange sehr gut ist, bis nicht mehr als 0,5 Substitutionen pro Base stattgefunden haben. Bei dem Vorgänger von andi, dem Programm kr [9], führten Schwankungen in der Mutationsrate entlang von Sequenzen zur Unterschätzung des wahren Abstands. Solche Schwankungen kommen in der Natur oft vor, zum Beispiel durch Rekombination im Zuge der Meiose. Daher ist ein großer Vorteil von andi, dass seine Genauigkeit dadurch nicht beeinflusst wird [4].
Neben der Genauigkeit ist außerdem geringer Ressourcenbedarf in der IT-Technik gefordert. Der Speicherplatzbedarf von andi beträgt etwa 45 Bytes pro Base, wobei nur Platz proportional zur längsten Sequenz verbraucht wird, da immer nur das Suffix-Array einer Sequenz sich im Speicher befindet. Was den Zeitbedarf angeht, so verläuft die Ankersuche mit Suffixbäumen bereits sehr schnell. Die Geschwindigkeit von andi ist weiter durch Parallelisierung bis maximal um einen Faktor beschleunigt, der gleich der Zahl der verfügbaren CPUs ist. Außerdem berechnet andi häufig wiederholte Suchabfragen im Voraus. Die Endgeschwindigkeit ist ungefähr eine Sekunde pro Megabase eines zu vergleichenden Sequenzpaares. Idealerweise wäre diese Geschwindigkeit unabhängig von der Gesamtlänge der Sequenz, was bei andi zwar nicht ganz, aber annähernd der Fall ist.
Große Phylogenien


Am 11. Dezember 2014 enthielt die zentrale europäische Sequenz-Datenbank ENSEMBL 2010 Escherichia coli Genome, die jeweils zwischen vier und sechs Millionen Basen lang sind. Viele dieser Genome sind nicht vollständig durchsequenziert, sondern bestehen aus hunderten von ungeordneten Fragmenten. andi ist nicht darauf angewiesen, dass ein Genom vollständig sequenziert ist, und rechnete in knapp zehn Stunden aus den E. coli Genomen den Baum in Abbildung 4 aus. Der dafür verwendete Computer hat 32 CPUs und 20 GB freien Zwischenspeicher (RAM), was heutzutage in der Forschung nichts Besonderes ist. Da sich viele E. coli Stämme sehr ähnlich sind, gibt es in Abbildung 4 hauptsächlich nur kurze Äste, aber ein paar fallen aus dem Rahmen, insbesondere die mit dem Pfeil markierten am obersten Rand. Durch Vergleich eines kleinen Stücks ihrer Genome mit allen anderen bekannten Bakteriengenomen stellte sich heraus, dass es sich gar nicht um E. coli, sondern um Klebsiellen handelt. Diese Bakterien kommen typischerweise im Darm zusammen mit E. coli vor.
Hier findet sich also eine weitere Anwendung für Phylogenien: Qualitätskontrolle für die immer schneller wachsenden Genomsammlungen. Damit zeigt sich andi als ein Werkzeug, das Biomedizinern hilft, auch in Zukunft Schritt zu halten mit der erstaunlichen Verfügbarkeit von Genomsequenzen.
Literaturhinweise
Bioinformatics, Advance Access
DOI: 10.1093/bioinformatics/btu815
In: Society for Industrial and Applied Mathematics - Proceedings of the first annual ACM-SIAM Symposium on Discrete Algorithms, SODA '90, 319-327 (1990); Philadelphia, PA, USA,
Selbstverlag (2013)
ISBN: 978-3-00-041316-2