Wie man durch Modelle die Entstehung des Erbguts und die Bildung von Arten beschreiben kann
Forschungsbericht (importiert) 2015 - Max Planck Institut für Evolutionsbiologie
Wie man Genome in Populationen modelliert
Ein Genom enthält die gesamte für die Entwicklung eines Individuums benötigte Information. Es stammt aus dem jeweiligen Erbgut seiner Eltern, unterscheidet sich aber wegen zweier grundlegender Mechanismen, die unweigerlich mit sexueller Fortpflanzung verbunden sind: Mutation und Rekombination. Während Mutationen in Keimzellen zufällige Veränderungen einbauen, verbindet die Rekombination zufällig Teile des jeweils ursprünglichen Erbguts der Eltern miteinander. Da sich diese Unterschiede mit der Zeit ansammeln, sind die Genome verwandtschaftlich nahestehender Personen ähnlicher als diejenigen entfernt verwandter Individuen. Genome enthalten daher auch Informationen über ihre eigene Entstehungsgeschichte. Man kann somit die Entstehung der Arten auch aus dem Grad der Ähnlichkeit ihres Erbguts ableiten.
Populationsgenetik untersucht, wie Gene innerhalb der Bevölkerung entstehen. Biologische Modelle können die Entstehung von Genen und Allelen in einer oder mehreren Populationen über die Zeit beschreiben. Als Grundlage hierfür dienen Mutationen, Rekombinationen und der Vorteil, der genau diese neue Genvariante seinen Trägern verschafft - auch relative Fitness genannt. Die Anzahl der Nachkommen eines Individuums, die als eine Komponente seiner Fitness zählt, ist aber nicht vollständig durch dessen Gene bestimmt, sondern sie erfolgt zu einem großen Teil auch zufällig, um Beispiel durch Partnerwahl, historische Aspekte der gegebenen Population, etc.. Somit wird die Evolution von Genen und letztlich des gesamten Erbguts am Ende von zufälligen Effekten bestimmt, die auch als Gendrift bezeichnet werden. Die einfachsten Modelle für die Genomentstehung in der Bevölkerung müssen daher drei Mechanismen berücksichtigen: Mutation, Rekombination und Gendrift.
Die sogenannte Koaleszenztheorie beschreibt auf mathematische Weise, wie einzelne Gene verschiedener Individuen miteinander verwandt sein können. Es ist eine große Herausforderung, diese Modelle auf vollständige Genome anzuwenden, da der Prozess der Rekombination das Erbgut in ein Mosaik von Segmenten mit unterschiedlichen, aber nicht unabhängigen Hintergrundinformationen zerstückelt. Mehrere klassische Modelle wurden dahingehend erweitert, dass die Entwicklung genomweiter Sequenzen in Populationen oder eng verwandter Arten nun unter dem Einfluss von Rekombination dargestellt wird. Um die Berechnungen vereinfachen zu können, basieren diese Erweiterungen alle auf einer gemeinsamen Schätzung, inwieweit die Rekombination das Genom beeinflusst. Der Prozess, der sich daraus ergibt, wird Markov-Kette genannt: An jeder beliebigen Stelle des Genoms hängt die zugrunde liegende lokale Hintergrundinformation von den Verwandtschaftsverhältnissen der umliegenden Stellen ab. Somit kann man ein sehr praktisches und gut untersuchtes mathematisches Modell namens Hidden Markov Modell (HMM) anwenden, um die Gesamtheit der Rekombinationen eines Genoms darzustellen, die von außen betrachtet verborgen (hidden) ist. Ein erweiteres Modell, das sequentially Markov coalescent (SMC) [1] oder Coalescent Hidden Markov Modell (CoalHMM) [2] genannt wird, kann daraus abgeleitet werden.
Wie man die Stammesgeschichte aus dem Erbgut heutiger Arten ableiten kann
Der CoalHMM-Ansatz wurde auf eng miteinander verwandte Arten, wie Mensch, Schimpanse und Gorilla, angewendet. Diese Ansätze erlauben, zusätzlich zur Ableitung der Entstehungsgeschichte, die wir mit Schimpansen und Bonobos teilen [3], den Rekombinations-Stammbaum (ancestral recombination graph, ARG, Abb. 1) umzustellen. Dieser verdeutlicht die lokalen Verwandtschaftsverhältnisse jeder einzelnen Position im Erbgut sowie einen großen Teil der Rekombinationsereignisse, die in der Vergangenheit aufgetreten sind. Beim Vergleich von eng miteinander verwandter Arten tritt im Rekombinations-Stammbaum ein bestimmter Mechanismus auf, den Fachleute das incomplete lineage sorting (ILS) nennen: Manche DNA-Bereiche können sich am ähnlichsten sein, beispielsweise bei Mensch und Gorilla,, obwohl sie nicht zu den am engsten verwandten Arten gehören, hier: Menschen und Schimpansen.
, der kurze Genomsegmente zeigt, in denen zwei Rekombinationsereignisse stattgefunden haben (R1 und R2). Bei den Arten handelt es sich um Mensch (H), Schimpanse (C) und Gorilla (G). Der senkrechte Pfeil gibt die Zeit an, von der Gegenwart in die Vergangenheit. Unten: Die farbigen Felder repräsentieren die drei Genomregionen mit ihren entsprechenden Stammesgeschichten. Der ARG enthält alle Informationen der lokalen Verwandtschaftsverhältnisse der DNA sowie die Zeit und den Ort, an denen die Rekombination anhält. Der mittlere, rote Graph zeigt das incomplete lineage sorting (ILS), da die beiden untersuchten Sequenzen sich zwar am ähnlichsten sind, aber zu zwei Arten gehören, nicht allzu nah miteinander verwandt sind.")
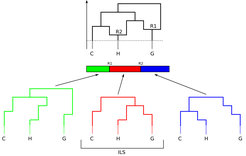
Die Möglichkeit, den Rekombinations-Stammbaum und die Struktur des incomplete lineage sorting umstellen zu können, ist eine wesentliche Errungenschaft, weil es Aufschlüsse über die evolutionären Prozesse gibt, die in ursprünglichen Arten aufgetreten sind - darunter auch der Zeitpunkt, in dem sich die Arten Mensch und Schimpanse aufgespalten haben. Der Rekombinations-Stammbaum macht es auch möglich, Rückschlüsse auf die Rekombinationsmuster in der Stammart zu ziehen. Eine solche rekonstruierte Übersicht von Rekombinationsmomenten in den Vorfahren kann nun mit denen von heutigen Arten verglichen werden, um so die Entwicklung der Rekombination im Genom zu untersuchen. Im Vergleich zeigt sich, dass sich die Rekombinationsmomente im Menschen schneller entwickelt haben als im Schimpansen [4].
Bestimmte Kräfte können den Rekombinations-Stammbaum allerdings verfälschen. Das kann dazu führen, dass die lokale Genvielfalt kleiner wird - oder aber in einigen Fällen viel zu groß wird. Zwei selektive Mechanismen sind dafür bekannt, sich auf die Vielfalt des Genoms auszuwirken. Schädliche Mutationen werden aus Populationen entfernt, da ihre Träger keine oder nur wenige Nachkommen haben; ein Phänomen, das als natürliche Selektion bezeichnet wird. Natürliche Selektion betrifft nicht nur die DNA-Stellen, in denen die Mutation auftritt. Es werden auch Allele, die mit ihnen verknüpft sind, entfernt. Die genetische Vielfalt ist somit an solchen natürlich selektierten Positionen kleiner. Diesen Prozess nennen Fachleute background selection. Ein weiteres Phänomen tritt auf, wenn bestimmte Gen-Varianten bevorzugt werden, weil sie einen Vorteil für ihre Träger bringen. Sie können sich schnell in einer Population anreichern und ausbreiten, weil sie nahe gelegene Genabschnitte gerichtet verdrängen. Man spricht vom selective sweep (sweep für Auskehren oder Verdrängen).
Es ist bekannt, dass background selection in vielen ursprünglichen Arten Spuren im Genom hinterlässt [3, 5]. Vor kurzem wurde festgestellt, dass das X-Chromosom des gemeinsamen Vorfahren von Mensch und Schimpanse große Regionen enthält, in denen das incomplete lineage sorting verloren gegangen ist. In diesen Regionen ist das Erbgut von Mensch und Schimpanse fast immer ähnlicher als das von Mensch und Gorilla. Dies ist eine unerwartete Beobachtung, wenn man Gendrift als einzige treibende Kraft ansieht, weil die Aufspaltung von Mensch, Schimpanse und Gorilla nämlich dicht beieinander liegt (Abb. 2). Dennoch lässt sich daraus schließen, dass sich die genetische Vielfalt im gemeinsamen Vorfahren von Mensch und Schimpanse verkleinert hat.
 und asiatischen (ASN) Bevölkerungsgruppen, in denen keinerlei Überbleibsel des Neanderthaler-Genoms vorhanden ist, wie in [6] beschrieben. Daten und Methoden werden in [7] genauer beschrieben.")
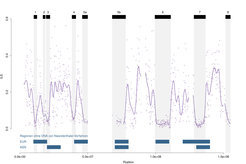
Das Fehlen des incomplete lineage sorting in so großen Regionen kann jedoch nicht allein durch genetische Drift erklärt werden. So zeigen die Autosomen ein wesentlich gleichmäßigeres Muster des incomplete lineage sorting mit vergleichsweise kleineren und weniger Regionen, die eine geringere Genvielfalt aufweisen. Demnach müssen selektive Kräfte in diesen Regionen offenbar eine große Rolle spielen. Es konnte errechnet werden, dass einzig und allein mit der grundlegenden Selektion das zu beobachtende Muster auf dem X-Chromosom nicht erklärt werden kann. In ähnlicher Weise zeigten Simulationen, dass ein einziger selective sweep den Verlust des incomplete lineage sorting in größeren DNA-Abschnitten ebenso nicht erklären kann. Es wird daher angenommen, dass wiederkehrende selective sweeps in jeder dieser Regionen des X-Chromosoms aufgetreten sein müssen.
Interessant hierbei ist, dass diese Regionen im X-Chromosom des gemeinsamen Vorfahren von Mensch und Schimpanse deutlich mit denjenigen untersuchten Regionen überlappen, die keinerlei Überbleibsel von Neanderthaler-DNA aufweisen ([6]; Abb. 2). Es wird daher vermutet, dass diese Regionen beteiligt waren, als sich reproduktive Barrieren zwischen unseren Vorfahren und denen der Schimpansen gebildet haben. Sie korrelieren auch mit DNA-Abschnitten in modernen menschlichen Populationen, die für eine geringere Vielfalt verantwortlich sind [7]. Viel überraschender aber ist, dass eine erheblich verkleinerte Genvielfalt auch an der gleichen Position in mehreren anderen Menschenaffenarten beobachtet werden kann [7, 8]. Das bedeutet, dass diese bestimmten Regionen auch eine Rolle gespielt haben, als sich weitere Primatenarten aufgespalten haben. Es ist nun eine große Aufgabe, die molekularen Mechanismen zu entschlüsseln, die an der Entstehung der selective sweeps beteiligt sind, um auch die Entstehungsgeschichte unserer eigenen Art und ihre Verwandtschaftsverhältnisse zu den Menschenaffen besser verstehen zu können. Aktuelle Hypothesen hierzu beschäftigen sich mit der gerichteten Selektion, darunter auch dem genetischen Unterschied zwischen den Geschlechtern [7].
Wie man unterschiedliche genomische Landschaften mit realistischen Bevölkerungsentwicklungen verknüpfen kann
Die Erstellung bisheriger Modelle war darauf konzentriert, komplexe Bevölkerungsentwicklungen einzubinden, um beispielsweise die Bevölkerungsstruktur und Zu- bzw Abwanderungen zu erklären [9]. Diese Modelle gehen jedoch davon aus, dass alle Kräfte gleichmäßig auf das Genom wirken, was ganz eindeutig im Widerspruch zu unserem derzeitigen Wissen über die Biologie der Erbsubstanz vieler Arten steht. Mutation, Rekombination und Selektion können ganz beträchtlich zwischen den einzelnen Regionen eines bestimmten Genoms variieren (Abb. 3).

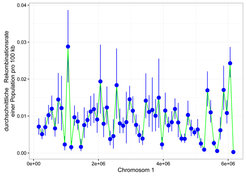
Daher ist es für das Berechnen von biologischen Modellen, die Genome in der Bevölkerung abbilden, eine neue Herausforderung, zusätzlich zu realistischen Bevölkerungsentwicklungen auch die unterschiedlichen genomischen Landschaften zu erklären. Neue Modelle, die solche Unterschiede berücksichtigen, sind aber eine Voraussetzung dafür, die Entwicklungsgeschichte vieler Organismen besser verstehen zu können und sind daher die primären Forschungsziele der Forschergruppe Molekulare System Evolution am Institut.