Forschung
Unsere Forschung konzentriert sich darauf, wie die genetischen Informationen von einer Generation zur nächsten durch meiotische Rekombination neu gemischt werden. Meiotische Rekombinationsereignisse sind nicht zufällig über das Genom verteilt, sondern konzentrieren sich in Rekombinationshotspots, welche etwa 1-2 kilo-basen lange Genomsequenzen sind. Das Protein PRDM9 bindet Motive in Rekombinationshotspots mit Hilfe seiner DNS Bindedomäne. Die DNS-Bindedomäne von PRDM9 besteht aus einer variablen Anzahl von C2H2-Zinkfingern, welche eine beschleunigte Evolution aufweisen. Die Folge der rapiden Evolution ist, dass Rekombinationslandschaften in verschiedener Spezies nicht, oder nur sehr gering, überlappen. Dies gilt sogar für engverwandte Arten wie Menschen und Schimpansen.
Die C2H2-Zinkfinger Domäne von PRDM9 zeigt auch eine hohe intra-spezifische Variation. Manch Kreuzungen von Mäusen führen zu männlicher Hybridsterilität in der F1-Generation, weil die elterlichen Kombinationen von Hotspot-Motiven sowie PRDM9-Varianten nicht miteinander kompatibel sind. Dies macht das PRDM9-Gen zu einem Speziations-Gen. PRDM9 hat auch eine SET-Domäne mit Histon-3-Lysin 4 (H3K4)- und Histon-3-Lysin-36-Trimethylierungsaktivität, mit denen PRDM9 die Histone in Hotspots für die Rekombinations-initiation markiert werden. Bei PRDM9-Knockout-Mäusen fehlt diese Markierung und die Rekombination wird stattdessen an existierenden Tri-methylierungs-markierungen initiiert, beispielsweise in Promotoren. Der Mangel an funktionellem PRDM9 führt dadurch zu meiotischem Arrest, die Konsequenz der resultierenden Azoospermie ist Unfruchtbarkeit.
Aktuelle Forschungsschwerpunkte
Der Mechanismus der Rekombination in Abwesenheit von PRDM9
")

Ganze Familien, wie die Säugetierfamilie der Canidae, zu der Hunde und Wölfe gehören, sowie mehrere Vogel- und Fischarten, verfügen über kein funktionelles PRDM9 Protein. Sie sind jedoch dennoch in der Lage, die Meiose erfolgreich abzuschließen. Die Genome von Hunden sowie Zebrafinken und Spitzschwanzamadine besitzen dennoch charakteristische Signaturen von historischer Rekombination, die als Stellen von genetischem Kopplungsungleichgewicht sichtbar sind.

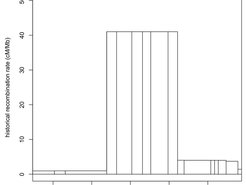
Wir erforschen nicht nur die Häufigkeit der Rekombinations-Ereignisse, sondern auch deren detaillierte Verteilung innerhalb dieser Rekombinations-Hotspots. Wir sind Experten darin, die Stellen der historischen Rekombination für aktive (de-novo) Rekombination zu untersuchen. Wir bedienen uns dabei verschiedener Spermien-Typisierungs-Ansätze. Ebenso verwenden wir Immunfluoreszenz-Färbung von meiotischen Proteinen (siehe Bild im Banner), um den zeitlichen Ablauf der Meiose sowie die Schwere möglicher Defekte wie meiotischem Arrest sowie Asynapse zu bestimmen.
Unser Ziel ist es detaillierte Rekombinationsmuster von Tieren mit PRDM9-unabhängiger Hotspotregulation mit denen von Organismen die PRDM9-regulierte Hotspots besitzen, zu vergleichen. Dies liefert detaillierte Einblicke in die Unterschiede der Dynamik und der Kontrollfaktoren von Rekombination in Abwesenheit von PRDM9, und trägt auch zu einem besseren Verständnis der Funktion und der Rolle von PRDM9 bei normaler Meiose bei. Dazu untersuchen wir zahlreiche Proben von PRDM9-defizienten Organismen, darunter mehrere Arten der Familie der Canidae sowie Singvögel. Diese vergleichen wir mit den Rekombinationsmustern an PRDM9 regulierten Hotspots, vor allem von mehreren Arten und Unterarten von Mäusen der gesamten Gattung Mus, einschliesslich Mus musculus musculus, Mus musculus domesticus, Mus spretus und Mus spicilegus sowie Apodemus uraliensis als “Outgroup”.
Die Evolution von Rekombinationslandschaften

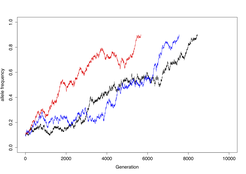
Rekombinationslandschaften verändern sich rasch. PRDM9-regulierte Hotspots haben Rekombinationsinitiations-Motive, welche die Bindungsstellen für PRDM9 sind. Mutationen in diesen Motiven führen zur Inaktivierung der Bindungsstelle. In Individuen die heterozygot für die inaktivierende Mutation sind, sorgt Genkonversion überschreibt das aktive Motiv mit einem weniger aktiven, oder gar inaktivem, Motiv. Über viele Generationen hinweg führt das wiederkehrende Überschreiben von aktiven Motiven in inaktive Motive schließlich zu einer kompletten Abschaltung eines Rekombinations-Hotspots in der gesamten Population.
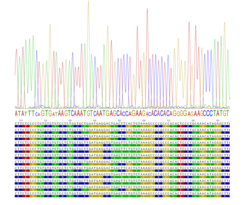
Zusätzliche Segregations-Distorsionen können durch GC-Basen-bevorzugende Genkonversion entstehen. Auch wenn diese die Crossover- oder Hotspot-Aktivität nicht beeinflussen, verändern sie die Genomkomposition über Generationen hinweg. Um das Auftreten von bevorzugender Genkonversion, sowie die Schwere von möglicher Segregations-Distorsion, aufzudecken, analysieren wir beide reziproken Rekombinationsrichtungen getrennt voneinander. Wir verwenden hierzu Allel-spezifischer PCR-Ansätze, die für beide Haplotypen spezifisch sind. Damit können wir die Häufigkeit und die Verteilung der reziproken Rekombinationsereignisse vergleichen. Diese Ergebnisse integrieren wir in Genom-evolutions-Modelle um den Hotspot-Umsatz und die Sequenz-Evolution zu simulieren, sowie deren langfristigen evolutionären Konsequenzen aufzudecken.